14.12.2012 | 15:23
Primary Upper Motor Neuron Disorders Chart
Primary Upper Motor Neuron Disorders Chart Reviewed by John K. Fink, M.D. , SPF Medical Advisor
><<>>< |
 Vísindi og fræði | Breytt 17.12.2012 kl. 08:49 | Slóð | Facebook
Vísindi og fræði | Breytt 17.12.2012 kl. 08:49 | Slóð | Facebook
11.12.2012 | 18:13
Astrocytes - hatursfullt árásarlið eða fórnfúsir verjendur ?
Astrocytes - hatursfullt árásarlið eða fórnfúsir verjendur ? Birtist fyrst 07. október 2011.
Efri og neðri hreyfitaugar. Loftur Altice Þorsteinsson.Hreyfitaugar liggja frá hreyfisvæðinu (primary motor cortex) á heilaberki manna, niður mænuna, þar sem aðrar hreyfitaugar taka við boðum sem berast út til vöðvanna. Hreyfitaugarnar lifa í sambýli við nokkrar aðrar frumur (límfrumur = glial cells), sem fram að þessu hefur verið álitið að væru ávallt vinsamlegir nágrannar. Nú hafa rannsóknir leitt í ljós að ein tegund límfruma á það til að færa hreyfitaugunum banabita. Þessar límfrumur nefnast Astrocytes (stjörnufrumur). Ef Astrocytes skipta skapi getur afleiðingin orðið hreyfitaugahrörnum (Motor Neuron Disease = MND). Stjörnufrumur (Astrocytes) eru vel tengdar. Við eðlilegar aðstæður annast Astrocytes margvísleg mikilvæg verkefni til að viðhalda eðlilegri starfsemi taugakerfisins. Astrocytes stjórna efnaskiptum (metabolism) taugafruma og flæði næringarefna (trophic maintenance) til þeirra. Einnig annast Astrocytes varnir gegn sýkingum í taugakerfinu (ásamt Microglia). Astrocytes eru algengustu frumur taugakerfisins og til dæmis 40% fleirri en taugafrumurnar sjálfar.  Sambúð frumanna í taugakerfinu er oftast góð. Hreyfitaugahrörnun (MND) er gjarnan skipt í ættgengt MND (FMND = Familial MND) og einstaklingsbundið MND (SMND = Sporadic MND). Um 10% MND tilfella eru ættgeng og fram að þessu hafa rannsóknir einkum beinst að þeirra tegund. Í þessu sambandi hefur mesta athygli vakið stökkbreitt enzyme sem nefnist SOD1. Stökkbreytt SOD1 hefur fundist í 20% tilfella af FMND, þannig að álitið er að einungis 2% allra MND tilfella séu með stökkbreytt SOD1. Nú berast þær fréttir að rannsóknir sýni að bæði FMND og SMND stafi af eitrun frá SOD1 og að sú eitrun komi eingöngu frá Astrocytes. Til dæmis valdi SOD1 í taugafrumunum sjálfum engum skaða. Frá þessum merku niðurstöðum er greint í ágúst-hefti Nature Biotechnology (Brian K. Kaspar og fl.), undir nafninu: Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Heldstu niðurstöður greinarinnar:
 Ættartré hreyfitaugar (Neuron) og Astrocyte. Hvað er SOD1 ? SOD stendur fyrir Superoxide Dismutase, sem er enzyme sem finnst inni í frumum manns-líkamans og breytir superoxíði (O2- ) í vetnis-peroxíð (H2O2) og súrefni (O2). Af SOD eru til þrjú afbrigði, með mismunandi málm-atómum: SOD1: Kopar, Zink - Superoxide Dismutase. SOD2: Mangan - Superoxide Dismutase. SOD3: Járn - Superoxide Dismutase. Efnabreytingunni er hægt að lýsa samkvæmt efnajöfnunni: Superoxide Dismutase 2 O2- + 2 H+ ààààààà H2O2 + O2 Önnur enzym eins og til dæmis Catalase eða Glutathione Peroxidase (GPx) taka síðan við verkinu og breyta peroxíðinu (H2O2) í skaðlaus efnasambönd vatns (H2O) og súrefnis (O2).  Hafa horfur á lækningu batnað ? Hægt er að fullyrða að með niðurstöðum þeirra rannsókna sem hér hefur nokkuð verið greint frá, hafa líkur aukist verulega að orsakir MND verði bráðlega uppgötvaðar. Nú þegar blasa við möguleikar til lækninga, en auðvitað getur framtíðin ein skorið úr um hvernig til mun takast. Einnig virðast þær aðferðir sem vísindamennirnir notuðu opna einnig möguleika til að skilja betur ýmsa aðra sjúkdóma, eins og Parkinson og Alzheimer. Eldri greinar mínar um MND: 08.04.2010: http://altice.blogcentral.is/blog/2010/10/9/hreyfitaugalomun-mnd-og-elliglop-ftd/ 04.07.2010: http://altice.blogcentral.is/blog/2010/10/10/fjogur-afbrigdi-hreyfitaugalomunar/
Greinar sem Brian K. Kaspar og félagar vísa til:
><<>>< |


Vísindi og fræði | Breytt 14.12.2012 kl. 15:31 | Slóð | Facebook
26.11.2012 | 21:15
Núna er rétti tíminn til að hefja MND-rannsóknir á Íslandi
Núna er rétti tíminn til að hefja MND-rannsóknir á Íslandi. Birtist fyrst í Morgunblaðinu 10. desember 2011.
Loftur Altice Þorsteinsson. Hreyfitaugasjúkdómar eru í flestum tilvikum banvæn mein, sem ekki hefur ennþá tekist að greina fullkomlega og sem engin lækning hefur fundist við. Sjúklingar með hreyfitaugasjúkdóma lamast oftast vegna hrörnunar hreyfitauganna, sem skiptast í efri og neðri hreyfitaugar. Efri hreyfitaugar liggja frá heilaberki niður mænuna, en neðri hreyfitaugar liggja frá mænu og heilastofni út í vöðva. Hver hreyfitaug er ein fruma og getur verið allt að metri á lengd. Fólk sem þjáist af þessum sjúkdómum lifir að meðaltali þrjú til fimm ár, frá því að skýr einkenni sjúkdómsins koma fram. Dánarorsök er oftast köfnun vegna lömunar þindarinnar eða vegna lungnasjúkdóma. Einungis um 10% sjúklinga hafa meira en 10 ára lífslíkur. Samkvæmt áætlunum frá WHO munu sjúkdómar í taugakerfinu hafa tekið við af krabbameinssjúkdómum árið 2040, sem önnur algengasta dánarorsök í efnahagslega velmegandi ríkjum og þar á meðal Íslandi. ><<>><
><<>>< |
26.11.2012 | 14:51
Fjögur afbrigði Hreyfitaugalömunar
Fjögur afbrigði Hreyfitaugalömunar
Fyrst birt 04. júlí 2010.
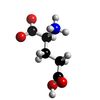
Glutamic acid
Loftur Altice Þorsteinsson. Hreyfitaugalömun (MND = Motor Neuron Disease) er skipt í fjögur afbrigði. Skiptingin ræðst af í hvoru hreyfitauga-kerfi líkamans sýkinguna er að finna. Hreyfitaugarnar eru í grófum dráttum frá heila til mænu, nefndar UMN (Upper Motor Neurons) og frá mænu til útlima, nefndar LMN (Lower Motor Neurons). Hreyfitaugalömunin getur birtst í öðru hreyfitauga-kerfinu, eða þeim báðum. Þótt hreyfitaugalömun sé ávallt alvarlegur sjúkdómur og erfiður, er munur á honum eftir afbrigðum og eftirfarandi flokkun segir nokkuð til um einkenni og lífslíkur.
• Amyotrophic lateral sclerosis (ALS) hrjáir 65% sjúklinga og er sjúkdómurinn í bæði UNM og LMN. Karlar eru um 2/3 sjúklinga, einkum yfir 55 ára aldri. Einkennin eru máttleysi, stýfleiki í vöðvum, sterk hreyfitauga-viðbrögð, tilfinningalegt ójafnvægi, vöðvakippir og þyngdar-tap. Að meðaltali lifa sjúklingar með þetta afbrigði tvö til fimm ár, frá því að sjúkdóms-einkenni koma fyrst í ljós.
• Progressive bulbar palsy (PBP) hrjáir 25% sjúklinga, einkum kvennfólk. Oft birtist sjúkdómurinn í bæði UMN og LMN, en þó takmarkaður við höfuð og háls. Þetta afbrigði einkennist af erfiðleikum við framburð orða (dysarthria) og erfiðleikum við að kyngja fæðu (dysphagia). Ef LMN er skaddað, birtast afleiðingarnar í nef-framburði máls, uppgangi fæðu um nef, tungu-rýrnun, vöðva-kippum og máttleysi hálsvöðva. Ef sjúkdómurinn birtist í UMN, eru afleiðingarnar stirðleiki í tungu, framburðar-hrinur og tilfinningalegt ójafnvægi. Líftími er oftast hálft ár til þrjú ár, frá því að einkenni birtast.
• Progressive muscular atrophy (PMA) hrjáir innan við 10% MND sjúklinga og leggst fyrst og fremst á unga karla. Orsökin er aðallega vegna LMN hrörnunar, sem leiðir til minnkandi styrks og rýrnunar vöðva, þyngdartaps og vöðvakippa. Að meðaltali lifa þessir sjúklingar fimm ár eða lengur. • Primary lateral sclerosis (PLS) hrjáir um það bil 2% MND sjúklinga. Tíðni þessa afbrigðis er tvöfalt meiri hjá körlum en kerlum og einkenni koma venjulega í ljós eftir 50 ára aldur. Einungis UMN hreyfitaugarnar eru skemmdar, sem leiðir til minnkandi styrks vöðva, stirðleika í liðum og harðari taugaviðbragða. PLS hefur ekki áhrif á lífslíkur manna.
Aðgreining á milli þessara afbrigða er oft erfið. Þegar sjúkdómurinn ágerist, sýnir reynslan að upphaflegar skilgreiningar reynast ónákvæmar, sem lýsir sér í rýrnun flestra vöðva og miklu máttleysi. Í flestum tilvikum (95%) er engin þekkt ástæða fyrir hreyfitaugalömun, gjarnan nefnt tilfallandi MND (sporadic MND). Þau 5% tilvika sem ekki eru ótengd, eru gjarnan nefnd ættgengd MND (familial MND) og eru sjúklingar þá erfðafræðilega tengdir, þannig að börn sjúks foreldris erfa til dæmis sjúkdóminn í 50% tilvika. Enginn munur á sjúkdóms-einkennum hefur verið greindur á milli tilfallandi MND og ættgengra MND.
Glutamic sýra ( C5H9NO4 ) => glutamate salt
Riluzole er eina lyfið sem hlotið hefur viðurkenningu til meðferðar á MND og eingöngu við ALS-afbrigðinu. Það er talið lengja líf sjúklinga um tvo til fjóra mánuði. Margir telja samt að Riluzole gagnist einnig gegn öðrum afbrigðum. Riluzole takmarkar framleiðslu líkamans á glutamate, en það efni örvar frumur í hreyfitaugum. Of mikið glutamate er talið skaðlegt fyrir hreyfitaugar og stuðla að skemmdum á þeim. Þetta er þó bara ein möguleg orsök fyrir MND.
• Glutamate er salt af Glutamic-sýru (H-C5H8NO4), sem myndast getur með málmum og stöðugum sameindum eins og Ammonium (NH4). Glutamic-sýra er ein af tuttugu Amino-sýrum líkamans og ein af þeim sem líkaminn framleiðir sjálfur. Hér er upptalning nokkurra glutamate salta:
><<>>< |



Vísindi og fræði | Breytt 14.12.2012 kl. 15:31 | Slóð | Facebook
25.11.2012 | 09:10
Information about ICRon-MND
International Center for Research on Motor Neuron Diseases(ICRon-MND)Registered in Iceland, under license number 561112-0960E-mail: hlutverk@simnet.isThe International Center for Research on Motor Neuron Diseases (ICRon-MND) is a non-profit organisation, located in Reykjavík, Iceland. It became a formal association in November 2012, created by individuals who recognized the need for increased research efforts to discover the causes of Motor Neuron Diseases.The aims of the International Center for Research on Motor Neuron Diseases are:1) To promote medical and scientific research into MND, in collaboration with Icelandic and foreign scientists and medical professionals.2) To raise funds for MND research, from official as well as private sources.3) To administer research grants for the study of MND.4) To create and exchange scientific knowledge about all aspects of MND.5) To promote exact genetic diagnoses of Icelandic MND patents.6) To undertake MND research projects.Board of the International Center for Research on Motor Neuron DiseasesLoftur Altice Þorsteinsson, MSc., MEd.Pétur Henry Petersen, PhD.Grétar Guðmundsson, MD.Páll Ragnar Karlsson, PhD. ><<>>< |
Vísindi og fræði | Breytt 14.12.2012 kl. 15:32 | Slóð | Facebook
24.11.2012 | 19:28
Greining á hreyfitauga sjúkdómum með rannsókn á erfðaefni
| |||||||||



| |
2002 | 2012 |
Framfarir í rannsóknum á erfðafræði ættgengs MND. | |


Á 10 ára tímabilinu 2002 – 2012 hafa greinst stökkbreytingar í mörgum genum MND-sjúklinga, sem ekki hafði áður verið vitað að tengdust MND. Árið 2002 voru 20% ættgengra MND-tilvika tengd ákveðnum stökkbreytingum, en árið 2012 voru 70% ættgengra tilvika tengd ákveðnum stökkbreytingum. Þessi þróun bendir til, að eftir önnur 10 ár verði allir MND-sjúklingar með örugga sjúkdóms-greiningu, sem byggir á rannsókn á erfðaefni þeirra og sem tengir sjúkdóminn við ákveðna stökkbreytingu í genum.

Gena-mengi frumanna er geymt í litninga-pörum (chromosome pairs) og í frumum manna eru 22 pör samlitninga og eitt par kynlitninga (X,Y). Litninga-pörin eru frá sitt hvoru foreldri og sama gildir því um genin, að þau koma hvort um sig frá föður og móður.

Algengasta erfða-mynstrið fyrir MND nefnist “jafnkynja ríkjandi” (autosomal dominant), sem merkir að stökkbreyting erfist jafnt til beggja kynja og að einungis eitt stökkbreytt gen þarf til að valda sjúkdómnum. MND-sjúklingur er því venjulega með eitt stökkbreytt gen og annað óbreytt. Barn foreldris með MND hefur 50% líkur að erfa hið gallaða gen og jafn miklar að erfa það ekki. Hins vegar er ekki öruggt að MND komi fram hjá einstaklingi, þótt hann sé með stökkbreytt gen, vegna þess að erfða-myndstur þess gens kann að vera annað en “jafnkynja ríkjandi”.
Leit að stökkbreyttum genum í próteinum sem tengd hafa verið MND, er því aðferð til að greina MND. Fram að þessu hefur leitin takmarkast við ættgeng afbrigði af MND, en sú mynd er að breytast. Jafnframt hafa flestar stökkbreytingar fundist í hvata (enzyme) sem nefnist SOD1 (Copper-Zink Superoxide Dismutase). Fundist hafa nær 200 stökkbreytingar í SOD1 og engin ástæða er til að ætla að leitinni sé lokið.
Í heilbrigðu ástandi er SOD1 mjög gagnlegt, því að það er kraftmikið andoxunarefni, sem breytir Superoxide í minna skaðlegt vetnis-peroxide (H2O2) og súrefni (O2). Önnur enzyme eins og til dæmis Catalase eða Glutathione Peroxidase (GPx) taka síðan við verkinu og breyta peroxíðinu (H2O2) í skaðlaus efnasambönd vatns (H2O) og súrefnis (O2).

SOD1-genið er samsett úr fimm svæðum sem nefnd eru exon. Stökkbreytingar sem tengjast MND hafa fundist í þeim öllum, en í mismunandi mæli. SOD1-G93S er eina stökkbreytta genið sem fundist hefur á Íslandi. Hér stendur G fyrir amino-sýru sem nefnd er Glycine, 93 er númerið á aðsetri (codon) breytingarinnar og S stendur fyrir aðra amino-sýru Serine. G93S merkir því, að á aðsetri 93 hefur orðið stökkbreyting, þannig að Glycine amono-sýra hefur breytst í Serene amino-sýru.

Afleiðingar stökkbreytinganna eru þær, að nytsamlegt SOD1 breytir um eðli og gefur frá sér eiturefni, sem er banvænt fyrir hreyfitaugarnar. Athyglisvert er, að það virðist vera stökkbreytt SOD1 í frumum sem nefnast stjörnufrumur (Astrocytes) sem gefur frá sér efni sem er banvænt fyrir hreyfitaugafrumur (Motor neurons), en ekki SOD1 í hreyfitaugafrumunum sjálfum.
Nokkrar aðferðir eru tiltækar til að greina stökkbreytingar í SOD1 og eru þær hver annari flóknari (virðist mér), nema High-Resolution Melting (HRM) (sem ég skil að einhverju leyti). Japanskur hópur vísindamanna hefur nýlega notað HRM til að greina stökkbreytingar í SOD1 hjá sjúklingum með tilfallandi MND.
Greining með HRM er fólgin í samanburði á ferlum “eðlilegs” og stökkbreytts SOD1. Skoðað er birtumagn sem fall af “bráðnunar”-hitastigi. Menn notfæra sér að ákveðin litarefni dofna við að erfðaefni klofnar við hitun.

Greining á G93S stökkbreytingu í SOD1 geni. Japan er eini staðurinn í heiminum þar sem SOD1-G93S hefur greinst, fyrir utan Ísland. Heimild: Mitsuya Morita, Japan.
><<>><
Eldri greinar mínar um MND:
10.12.2011: Núna er rétti tíminn til að hefja MND-rannsóknir á Íslandi.
07.10.2011: Astrocytes - hatusfullt árásarlið eða fórnfúsir verjendur ?
04.07.2010: Fjögur afbrigði hreyfitaugalömunar.
08.04.2010: Hreyfitaugalömun (MND) og Elliglöp (FTD).
><<>><
Vísindi og fræði | Breytt 3.4.2013 kl. 15:46 | Slóð | Facebook
Nýjustu færslur
- Opið bréf til Ban Ki-moon aðalritara Sameinuðu þjóðanna
- Að standa upp eftir byltu getur verið erfitt, eða ómögulegt !
- Rannsóknarklasi í hreyfitauga sjúkdómum er ekki lengur fjarlæ...
- Rannsóknir sýna að MND á Íslandi er arfgengur sjúkdómur
- Þingsályktun væntanleg um stuðning við MND-rannsóknir.
- Söfnunarreikningur MND-rannsókna
- Persónuvernd: Bréf til Íslendskrar erfðagreiningar vegna söfn...
- Therapy Slows Onset and Progression of ALS (Amyotrophic Later...
- Swelling of feet and legs - vandamál margra MND sjúklinga
- Clinical Spectrum of Motor Neuron Disorders
- Skýrslur: Primary Lateral Sclerosis
- Kári Stefánsson: Að bjarga eða bjarga ekki mannslífum
- DNA double helix: discovery that led to 60 years of biologica...
- Myriad Genetics CEO Claims He Owns Your Genes
- Erfðafræðibyltingin er tækifæri fyrir Ísland
Færsluflokkar
| Sept. 2025 | ||||||
| S | M | Þ | M | F | F | L |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| 7 | 8 | 9 | 10 | 11 | 12 | 13 |
| 14 | 15 | 16 | 17 | 18 | 19 | 20 |
| 21 | 22 | 23 | 24 | 25 | 26 | 27 |
| 28 | 29 | 30 | ||||